氧化铁纳米材料具有良好的生物安全性以及独特的磁学性能,在疾病的诊断治疗方面展现出巨大的应用潜力。然而,由于制备的氧化铁纳米材料粒径分布较宽、结晶度不足,限制了材料的临床转化与推广。在缺乏相关理论基础的前提下,仅依赖实验人员经验难以优化合成工艺,提高材料性能。虽然已有实验研究推测出多种晶体成核生长理论,但当前表征手段的空间/时间分辨率不够,无法实现对材料合成过程的直接观测。
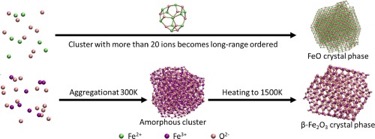
分子动力学模拟提供了一种数值方法来研究从自由离子到有序晶体的结构转变。然而,定义适当的力场参数对于氧化铁系统是一个重要的挑战。在这项研究中,我们通过考虑晶体结构中离子的力平衡,在多个初始参数的基础上拟合Coulombic-Buckingham的参数。我们的拟合力场根据以前发表的实验中获得的晶格常数和弹性特性进行了验证。在采用该力场的分子动力学模拟中,离子通过聚集产生了长程有序的FeO晶体结构。随着弛豫温度从600K提高到1500K,β-Fe2O3晶相从无定形或短程有序结构转变为ε-Fe2O3,这与已发表的实验结果很一致。
该论文“A force field for molecular dynamics simulations of iron oxide system”目前已被《Materials Science and Engineering: B》接收并在线发表: https://doi.org/10.1016/j.mseb.2022.115803。